Evaluating Genome Assembly Quality with QUAST: A Comprehensive Guide
Written on
QUAST, short for QUality Assessment Tool, is an essential resource for evaluating the quality of genome assemblies. As various assembly algorithms have been developed, it is crucial to choose the most suitable one for different scenarios to achieve optimal results. While contiguous genomes may not always be attainable, it is possible to derive segments from reference genomes using existing assembly methods, highlighting the importance of assembly quality assessments. These evaluations guide researchers in selecting the right assemblers based on their specific needs.
In this article, we will explore how to determine the quality of genome assemblies using QUAST, one of the leading assessment tools in the field.
What is QUAST?
QUAST allows users to evaluate assemblies with or without reference genomes and provides comprehensive reports, tables, and visualizations illustrating various assembly characteristics.
Downloading QUAST
To obtain QUAST, visit its official site and click the DOWNLOAD button. You will be redirected to a SOURCEFORGE page where you can download the latest version (as of this writing, it is quast-5.0.2). The pre-compiled binaries can be extracted and run immediately.
tar -xf quast-5.0.2.tar.gz cd quast-5.0.2 quast.py
After executing quast.py, you will see the following output:
QUAST: Quality Assessment Tool for Genome Assemblies Version: 5.0.2
Usage: python quast.py [options] <files_with_contigs>
Options include: - -o --output-dir <dirname>: Directory for saving result files [default: quast_results/results_<datetime>] - -r <filename>: Reference genome file - -g --features [type:]<filename>: File with genomic feature coordinates (GFF, BED, NCBI or TXT) - -m --min-contig <int>: Minimum contig length threshold [default: 500] - -t --threads <int>: Maximum number of threads [default: 25% of CPUs]
These are basic options; for a complete list, use --help. The online QUAST manual is available at http://quast.sf.net/manual.
Once QUAST is confirmed to be functioning correctly, we can proceed to assess some assemblies.
Obtaining an Example Assembly
We will utilize a sample dataset from the Flye assembler, which includes reads from an E. coli genome (Escherichia coli str. K-12 substr. MG1655, NCBI accession number CP009685). This dataset contains PacBio reads.
To download the dataset, use the following command: wget https://zenodo.org/record/1172816/files/E.coli_PacBio_40x.fasta
Next, we will assemble this dataset using the Flye assembler: flye --pacbio-raw E.coli_PacBio_40x.fasta --out-dir my_assembly --threads 8
The final assembly's contigs can be found in assembly.fasta. Let's check the quality of this assembly using QUAST.
Using QUAST
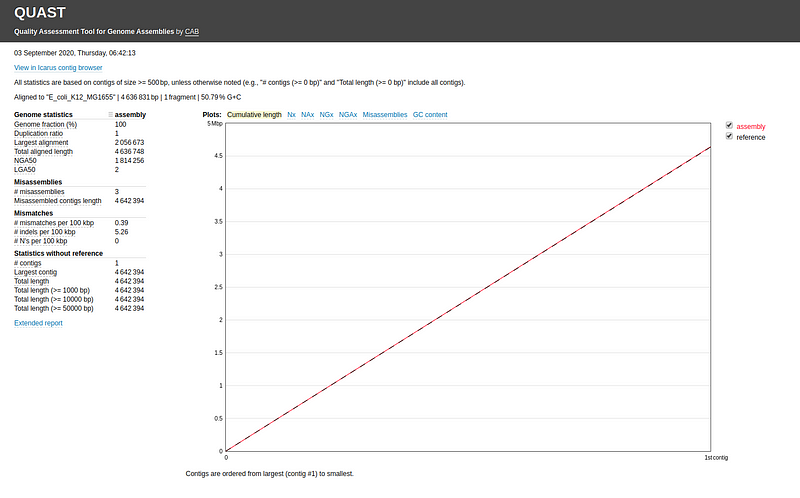
To run QUAST, provide the contigs file from the final assembly along with the reference genome: quast.py my_assembly/assembly.fasta -r ref.fasta -o quastResult
The report can be viewed in the report.html file located in the output folder.
You can also compare multiple assemblies (e.g., assembly1.fasta and assembly2.fasta) and assign labels for each: quast.py assembly1.fasta assembly2.fasta -l label1,label2 -r ref.fasta -o quastResult
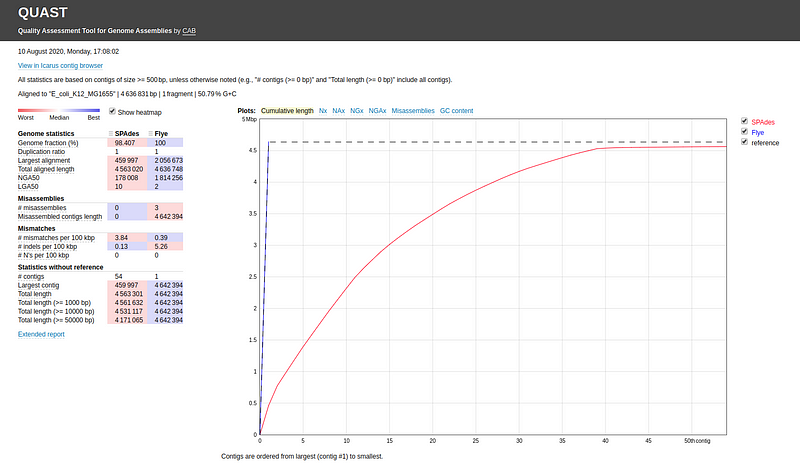
Common evaluation metrics include: - Genome fraction - Largest alignment - NGA50 - LGA50 - Number of misassemblies - Number of contigs
QUAST provides explanations for these metrics; hovering over each will display a popup with detailed descriptions.
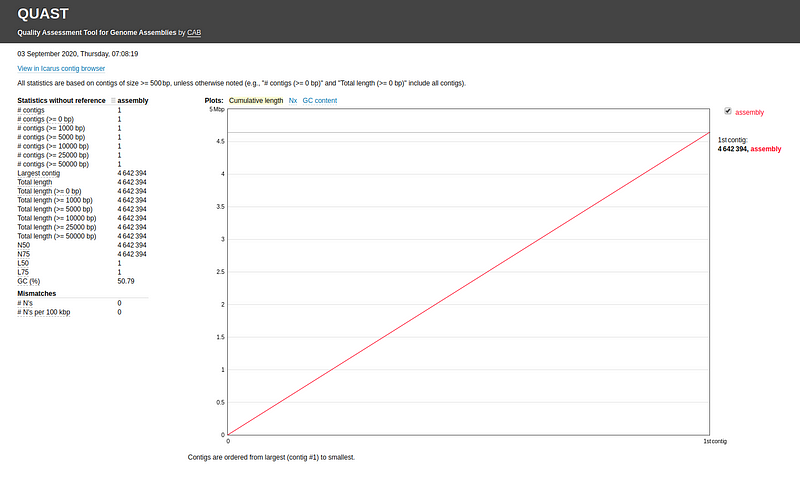
You can also evaluate your assembly without a reference genome: quast.py my_assembly/assembly.fasta -o quastResult
The results will provide statistics such as: - Number of contigs - Largest contig - Total length - N50 - L50
Icarus Contig Browser
Icarus is a visualization tool integrated within QUAST for analyzing assemblies.
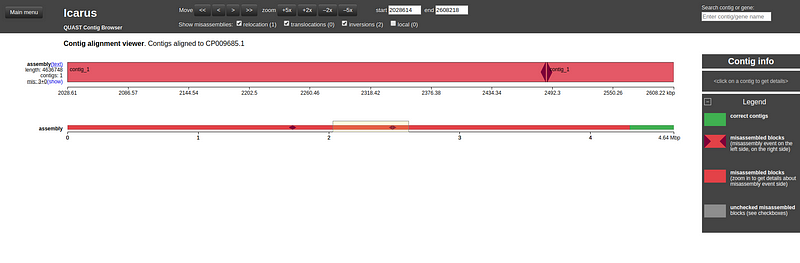
This tool allows you to see how closely your assembly aligns with the reference genome.
MetaQUAST: QUAST for Metagenomics Assemblies
QUAST also offers MetaQUAST, which is designed for assessing metagenomic assemblies. Users can compare multiple assemblies simultaneously and include several reference genomes.
To execute MetaQUAST, use the following command: metaquast.py meta.contigs1.fasta meta.contigs2.fasta -l label1,label2 -R References/ -t 8 -o metaquastResult
Similar to QUAST, labels can be assigned to each assembly for clarity in the final report. You can also specify a folder containing all reference genomes.
Final Thoughts
I hope this article serves as a valuable resource for understanding how to utilize quality assessment tools for genome assemblies. Feel free to incorporate these tools into your research and projects, as they are freely accessible.
Take care and stay safe!
For further reading, check out my previous articles on bioinformatics and DNA analysis.
<div class="link-block">
<div>
<div>
<h2>Bioinformatics and Computational Biology— What? Why? How?</h2>
<div><h3>A gentle introduction to bioinformatics and computational biology</h3></div>
<div><p>medium.com</p></div>
</div>
<div>
</div>
</div>
</div>
<div class="link-block">
<div>
<div>
<h2>A Dummies’ Intro to Bioinformatics</h2>
<div><h3>Bioinformatics is gaining traction in today’s scientific landscape.</h3></div>
<div><p>towardsdatascience.com</p></div>
</div>
<div>
</div>
</div>
</div>
<div class="link-block">
<div>
<div>
<h2>DNA Sequence Data Analysis</h2>
<div><h3>An introductory guide to DNA sequence data analysis.</h3></div>
<div><p>medium.com</p></div>
</div>
<div>
</div>
</div>
</div>
<div class="link-block">
<div>
<div>
<h2>Genome Assembly — The Holy Grail of Genome Analysis</h2>
<div><h3>Exploring the assembly of the 2019 novel coronavirus genome.</h3></div>
<div><p>towardsdatascience.com</p></div>
</div>
<div>
</div>
</div>
</div>
<div class="link-block">
<div>
<div>
<h2>A Simple Introduction to Read Simulators</h2>
<div><h3>An overview of read simulation tools and their applications.</h3></div>
<div><p>medium.com</p></div>
</div>
<div>
</div>
</div>
</div>